Introduction
The human body is made of trillions of cells, partitioned into hundreds of different types and subtypes that we can now characterize in high definition. Advances in library preparation and sequencing technologies have allowed the single-cell analysis community to investigate the content of each cell more accurately than ever. Currently, droplet sequencing is the primary method used to survey these cells, as it allows the capture of transcriptomes from thousands of cells in parallel and is useful for cell-type identification. However, since the droplet-based method focuses on single mRNA ends (typically the 3′ end), it gathers limited information. Complementary methods that provide full-length mRNA information, such as the Smart-seq2 method (Picelli et al. 2013) or Takara Bio’s SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (SSv4), are preferred for generating richer datasets. Using full-length and droplet-based methods in parallel is an emerging need in the scientific community, especially for consortia such as the Human Cell Atlas. There is still room for improvement of the Smart-seq2 method, particularly for use with challenging samples (e.g., cells with very low RNA content or nuclei). To address this need, we have further modified our core SMART-Seq technology to create a new chemistry with higher sensitivity designed specifically for single-cell applications—the SMART-Seq Single Cell Kit (Figure 1)—which outperforms all currently available full-length methods.
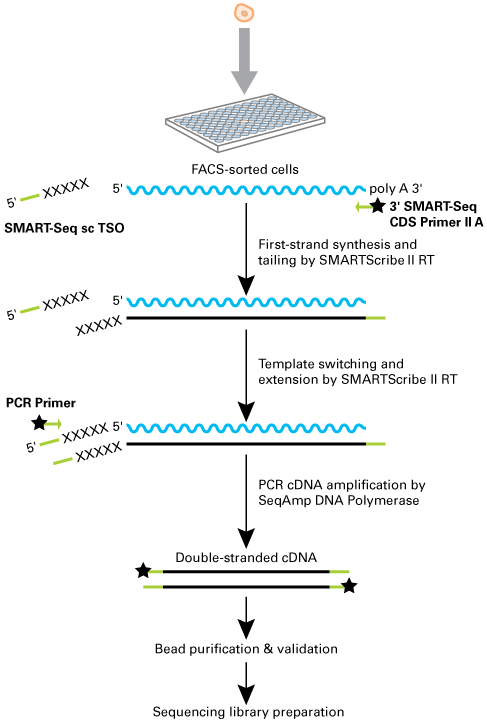
Figure 1. SMART-Seq Single Cell Kit technology and workflow. The SMART-Seq Single Cell Kit’s plate-based workflow allows direct input of single cells isolated by FACS or other methods. SMART technology (Chenchik et al. 1998) is then used in a ligation-free protocol to generate full-length cDNA. The reverse transcriptase (RT) adds nontemplated nucleotides (indicated by Xs) that hybridize to the SMART-Seq sc TSO, providing a new template for the RT. Chemical modifications to block ligation during sequencing library preparation (if using a ligation-based library preparation method) are present on some primers (indicated by the black stars). The SMART adapters, added by the oligo(dT) primer (3′ SMART-Seq CDS Primer II A) and SMART-Seq sc TSO, are indicated in green and used for amplification during PCR. The amplified cDNA is then purified, quantified, and used for sequencing library preparation (Illumina Nextera® XT kit).
Results
Greater sensitivity and reproducibility than Smart-seq2
The Smart-seq2 protocol (Picelli et al. 2013) and Takara Bio’s SMART-Seq technology are widely used by the scientific community to generate in-depth characterization of the transcriptome at the single-cell level. To compare performance between the new SMART-Seq Single Cell Kit (SSsc) and the Smart-seq2 protocol, we processed single cells from the lymphoblastoid cell line GM12878 according to each chemistry’s protocol (Figure 2).
We observed that the read distribution is different between the two chemistries. The Smart-seq2 chemistry had a much higher number of reads mapping to the mitochondrial genome (Figure 2, Panel A), resulting in a lower number of reads available for gene identification. We also observed a 15% increase in the number of genes identified in the cells processed with SSsc as compared to Smart-seq2 (Figure 2, Panel B). The higher sensitivity of the SSsc method is associated with greater reproducibility across cells, as indicated by the higher Spearman correlation within SSsc-processed cells (Figure 2, Panel C) and a lower dropout rate than Smart-seq2 (Figure 2, Panel D).
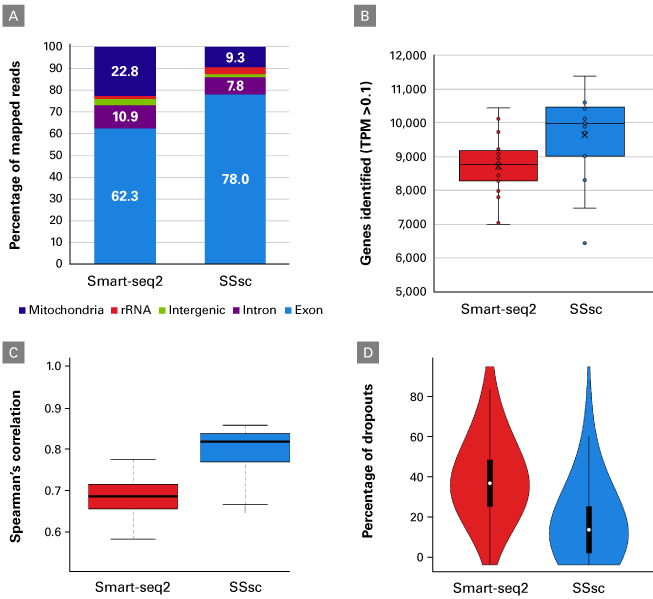
Figure 2. The SMART-Seq Single Cell Kit outperforms the Smart-seq2 protocol. Single cells from the lymphoblastoid cell line GM12878 were processed with SSsc (18 cells) or the Smart-seq2 protocol (20 cells; Picelli et al. 2014) using 19 cycles of PCR. As described in the methods, RNA-seq libraries were generated and sequences analyzed (after normalizing all samples to 1.75 million paired-end reads). Panel A. The read distribution varied between the two chemistries, with increased mitochondrial reads using Smart-seq2 and increased exonic reads using SSsc. Panel B. More genes were detected in the cells processed with SSsc. Panel C. Correlation boxplots showing the intragroup Spearman correlation between all cells processed with either method. The higher Spearman correlation among the cells processed with SSsc indicates a greater reproducibility than the Smart-seq2 method. Panel D. The greater reproducibility of SSsc is also demonstrated by the lower dropout rate of the genes detected with a TPM >1.
Greater sensitivity and reproducibility than SSv4 for single cells
The SSv4 kit is the most sensitive single-cell RNA-seq method available, and considered by many as the gold standard for plate-based full-length scRNA-seq (Hodge et al. 2019; Ibrayeva et al. 2019; and other references). Through several experiments, we were able to demonstrate that the new SSsc kit generates data with even superior sensitivity and reproducibility than the SSv4 kit.
For the first experiment, we used 2 pg of high-quality control RNA to compare the two kits (Table I). We observed a dramatically higher cDNA yield with the SSsc kit. When looking at three technical replicates, the SSsc kit generated on average a total of 13.1 ng of cDNA, while the SSv4 kit generated 6.7 ng, representing a 100% increase. Following sequencing and data analysis, we found that the SSsc kit identified about 15% more genes than SSv4. In addition, SSsc produced superior reproducibility, as evidenced by the increased Pearson and Spearman correlations (Figure 3). These scatter plots show that SSsc was able to detect additional genes that were expressed at a low level.
Table I. Increased sensitivity with the SMART-Seq Single Cell Kit. Replicate cDNA libraries were generated from 2 pg of Universal Human Reference (UHR) total RNA using the SMART-Seq v4 kit (SSv4) or the SMART-Seq Single Cell Kit (SSsc); all libraries were processed with 19 PCR cycles. As described in the methods, RNA-seq libraries were generated and sequences analyzed (after normalizing all samples to 1.6 million paired-end reads). SSsc identified about 15% more genes than SSv4.
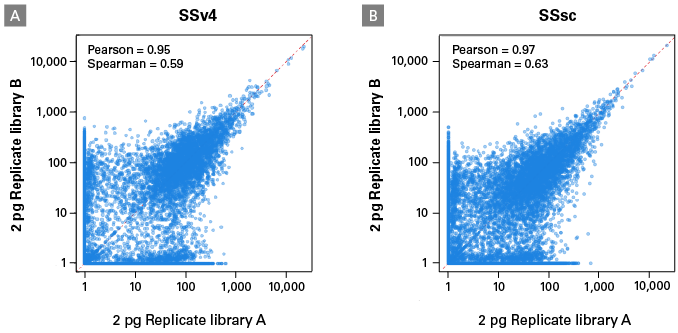
Figure 3. Increased reproducibility with the SMART-Seq Single Cell Kit. Libraries made from 2 pg of UHR total RNA (Table I) were analyzed using scatter plots to visualize the reproducibility between technical replicates (shown are TPM values from all genes with a log10+1 scale). SSv4 (Panel A) generated highly reproducible quantification, but SSsc (Panel B) produced superior reproducibility, as seen in the increased Pearson and Spearman correlations. In addition, SSsc was better at detecting low-expression genes.
In a second experiment, the performance of the SSsc kit was further evaluated using FACS-sorted cells. Single cells from lymphoblastoid cell line GM22601 (Figure 4) or the PBMC population from a healthy donor (Figure 5) were processed with SSv4 and SSsc. As seen in the first experiment using the UHR total RNA, the cDNA yield was dramatically higher with the SSsc kit (Figure 4, Panel A; data not shown for PBMCs). We continued to observe the SSsc kit’s improved performance in the sequencing data generated from both cell types. First, we observed that the read distribution was comparable between the two kits (Figure 4, Panel B and Figure 5, Panel A). Second, we found that we could identify more genes with the SSsc kit: ~50% increase in the GM22601 cell line (Figure 4, Panel C) and ~60% increase in the PBMC population (Figure 5, Panel B). This drastic increase in sensitivity with the SSsc kit held true over a wide range of sequencing depths (Figure 5, Panel B). For the relatively homogeneous cell population of the GM22601 line, the SSsc kit was more reproducible in terms of expression levels across all genes in the 12 cells analyzed, as shown by the higher Spearman correlation (Figure 4, Panel D)—in accordance with the data obtained using UHR total RNA (Table I).
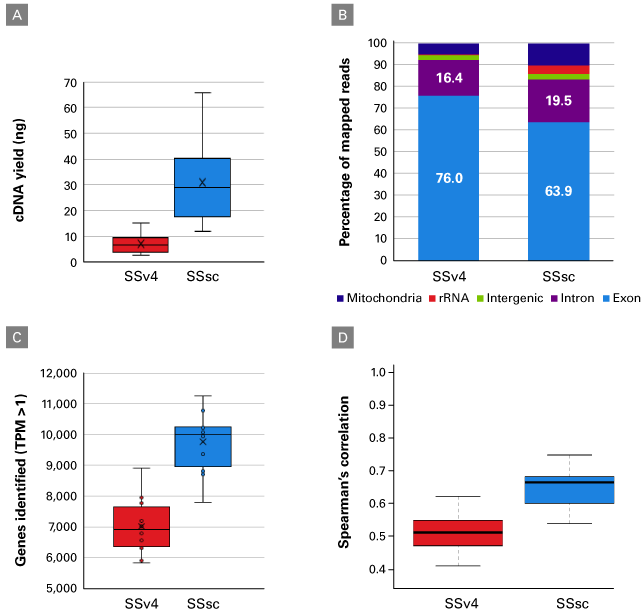
Figure 4. Improved performance for single cells with low RNA content. 12 single cells from lymphoblastoid cell line GM22601 were processed with SSv4 or SSsc using 19 cycles of PCR. As described in the methods, RNA-seq libraries were generated and sequences analyzed (after normalizing all samples to 1.25 million paired-end reads). Panel A. The cDNA yield generated with SSsc is drastically higher than that generated with SSv4. Panel B. The read distribution was fairly similar between the two chemistries. Panel C. Over 50% more genes were detected in the cells processed with SSsc. Panel D. Correlation boxplots showing intragroup Spearman correlation between all cells processed with either method. The higher Spearman correlation among the cells processed with SSsc indicates a greater reproducibility than SSv4.
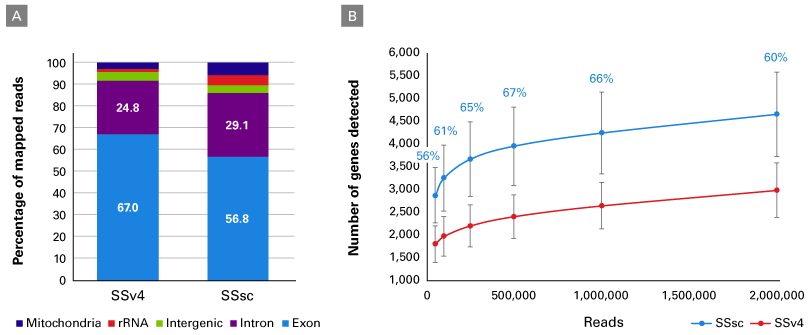
Figure 5. Improved performance with primary samples. PBMCs from a healthy donor were processed with the SSv4 or SSsc kit (~50 cells per kit). RNA-seq libraries were generated as described in the methods. Panel A. The read distribution is fairly similar between the two chemistries. Panel B. About 60% more genes are detected in the cells processed with SSsc, regardless of the number of reads used for the analysis.
Conclusions
Extracting meaningful biological information from the small amount of mRNA present in each cell requires an RNA-seq preparation method with exceptional sensitivity and reproducibility. To date, the SMART-Seq v4 kit has been the most sensitive commercial single-cell RNA-seq method, in part due to its incomparable capability to retrieve information from full-length mRNA and not just the 3′ end. To address the need for improvement with extremely challenging samples, such as cells with very low RNA content, we have further modified our core technology to create a new chemistry with higher sensitivity: the SMART-Seq Single Cell Kit. This kit outperforms all current commercial and noncommercial full-length methods, including Smart-seq2, against which our kit shines in terms of convenience, sensitivity, gene identification, and reproducibility. Added benefits are compatibility with automation platforms and a user-friendly plate-based workflow that starts directly from single cells isolated by FACS or other methods. In addition, the SMART-Seq single cell chemistry generates a high yield of cDNA, which is extremely useful when dealing with difficult cells such as clinical samples.
Methods
All cells were labeled with CD81-FITC antibody and 7-AAD (for distinguishing live from dead cells) prior to sorting using a BD FACSJazz cell sorter into a 96-well plate or PCR strips. For all comparison experiments, cells were sorted from a single batch of either GM12878, GM22601, or PBMCs. After sorting, cells were flash frozen on dry ice, and then stored at –80°C until ready to use. Unless otherwise noted, all libraries created with the SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing and the SMART-Seq Single Cell Kit were processed at full volume per the user manuals. For the comparison to the Smart-seq2 protocol, cells were sorted and processed as described in Picelli et al. 2014.
Sequencing libraries were generated using 125 pg of cDNA and the Nextera XT DNA Library Preparation Kit (Illumina) with a quarter of the recommended volume, as described in the SMART-Seq Single Cell Kit User Manual. Libraries were sequenced on a NextSeq® 500 instrument using 2 x 75 bp paired-end reads, and analysis was performed using CLC Genomics Workbench (mapping to the human [hg38] genome with Ensembl annotation). All percentages shown—including the number of reads that map to introns, exons, or intergenic regions—are percentages of mapped reads in each library.
References
Chenchik, A., Zhu, Y., Diatchenko, L., Li., R., Hill, J. & Siebert, P. Generation and use of high-quality cDNA from small amounts of total RNA by SMART PCR. In RT-PCR Methods for Gene Cloning and Analysis. Eds. Siebert, P. & Larrick, J. (BioTechniques Books, MA), pp. 305–319 (1998).
Hodge, R. D. et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019).
Ibrayeva, A. et al. Early Stem Cell Aging in the Mature Brain. bioRxiv 654608 (2019).
Picelli, S., Bjorklund, A. K., Faridani, O. R., Sagasser, S., Winberg, G., & Sandberg, R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098 (2013).
Picelli, S., Faridani, O. R., Bjorklund, A. K., Winberg, G., Sagasser, S. & Sandberg, R. Full-length RNA-Seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181 (2014).